The Clinical Trials Information System is a harmonised and simplified end-to-end single entry point for clinical trials information in the European Union and in the European Economic Area and will go live on 31 January 2022.
As a single entry point, it will be used for the submission, management and assessment of information throughout the life cycle of a clinical trial. The exchange of information between sponsors and Member States will be fully electronic.
In a recent EMA training webinar, the EMA advise the following to prepare for the CTIS go live date:
EMA has launched the CTIS training webpage containing a substantial amount of training materials and information for the users and organisations to facilitate their preparedness.
Should you need any support in clinical trial application just contact us and the Ivowen team will be here to help.
Written by Fiona Downey
https://eureg.ie/wp-content/uploads/2016/06/EMA.png297586ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2021-12-15 11:12:262023-11-06 11:08:28Are you ready for the Clinical Trials Information System (CTIS) implementation?
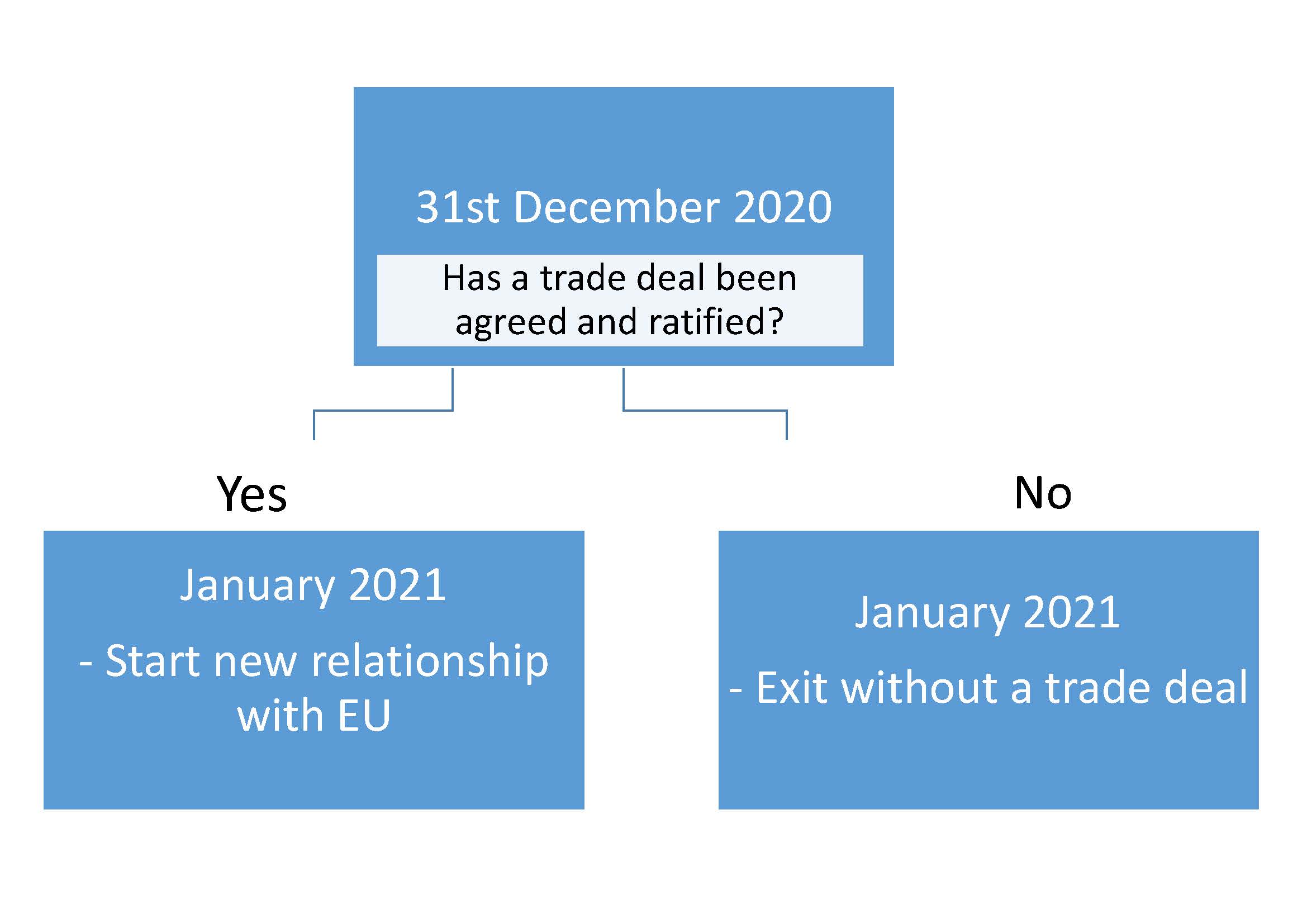
It seems that Brexit is on course for 1st January 2021….
The 30th June 2020 came and the legal deadline for agreeing to an extension of the transition period has passed with no request from the UK. Therefore the Brexit trade deal transition phase will come to an end on the 31st December 2020. At this stage there will be one of two possible outcomes:The next important dates are the 15-16th October (European Council Meeting) and 26th November (penultimate plenary session of 2020. European lawmakers have stated that a trade deal must be negotiated, checked, translated and presented to the European Parliament by this date if the transition period is to end by 31 December 2020). If the UK exits without a trade deal, trade between the UK and EU will change immediately on the 1st January 2021 (i.e. Hard Brexit).
The UK Government launched a major new public information campaign to give everyone the facts that they will need to be ready for 1 January 2021. A straightforward checker tool at gov.uk/transition will help identify some of the specific steps any business or individual needs to take to be ready, and will allow companies to sign up for bespoke updates.
If you require any assistance for UK products please contact us: info@eureg.ie
Written by Emily Fletcher
Emily Flecther
https://eureg.ie/wp-content/uploads/2020/07/Brexit-clock-image-22-07-20.jpg422750ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2020-07-22 12:42:222023-11-06 11:08:37BREXIT – No extension to the transition period
Did you know, that pharmaceutical companies can consult the European Medicines Agency (EMA) to determine whether a medicine they are developing is considered an advanced therapy medicinal product (ATMP)?
In doing so, they are allowed to receive confirmation that a medicine, which is based on genes, cells or tissues, meets the scientific criteria for defining an ATMP. The criteria for ATMPs are set out in Article 17 of Regulation (EC) No 1394/2007.
In order to submit an application for ATMP classification, applicants have to complete a:
pre-submission request form
ATMP-classification request form and briefing information
and return both forms to the EMA.
Applies to your company? Ivowen are here to assist you.
For more information on Ivowen’s services and how we can help you, contact us.
When the Clinical Trial Regulation (No. 536/2014) comes into effect in 2018, there will be a major change on how clinical trial applications are submitted and how clinical trials are conducted in the EU.
The goal of the new Regulation is to create an environment that is favourable to conducting clinical trials in the EU, with the highest standards of safety for participants and increased transparency of trial information.
Below you will find a brief summary of the changes:
Directive 2001/20/EC (Current)
Multiple submission for one trial (1 submission per each MS)
Double submission with a MSC: to NCA and EC
Individual assessment by Each MSC with no IT collaboration tool available
No Single MSC decision (NCA and ECs)
Limited EudraCT data availability to the public
Regulation 536/2014 (New)
Single e-submission to all MSCs
Harmonised dossier for one trial and
e-submission of structured data and documents by MSCs
Specific timeline
Joint assessment for PART I facilitated by collaboration tool
Single MSC decisions
Single web-based EU portal
Distribution of the burden among users
View all CT relation information
Transition period (3 years) Directive 2001/20/EC (current) to Regulation 573/2014
Starts when Regulation becomes applicable (~ Oct 2018)
1st Year: Clinical Trials can be submitted under old/current (Directive) or new (Regulation) system
2nd & 3rd Year: Trials authorised under old system can remain under that system, New/initial Clinical Trials should comply the Regulation System
All Clinical Trials to switch to the Regulation 3 years after implementation (~ Oct 2021)
Typical Documents to be submitted:
Part I: Cover letter (very important), EU AF, Protocol, IB, GMP compliance documents, IMPD, Auxiliary Medicinal Product Dossier, Scientific Advice, PIPs, and labelling, proof of payment, etc.
Part II: Recruitment arrangements, SI/ICF/ICF procedure, suitability of the investigator, suitability of the facilities, proof of insurance cover or indemnification, financial and other arrangement, proof of payment, etc.
Part I Timetable Coordinated assessment (also applies to Mono-national Clinical Trials)
Day 0: Validation
D26: Draft Part I Assessment Report made available by the RMS (reporting MS) to the CMS
D38: All CMS share considerations
D45: RMS finalises the Part Assessment Report
D57: Sponsor submits response (w/n 12days)
D69: Co-ordinated assessment between MSs (12 days)
D76: RMS files conclusion (7days)
Part II Timetable National evaluation
D0: Validation
D45: Final assessment report from each MSC submitted
D57: Sponsor submits response
D76: Final assessment by the MSC shall be performed (w/n 19 days)
Submission of Part I and II in parallel (recommended) or submission of Part I followed by Part II (not less than 2 years after Part I)
Important Notes:
New MSs can only be added after the notification date of the initial authorisation decision
Withdrawal of MS: the whole application has to be withdrawn and resubmitted
Transparency:
EU Database will be publically accessible by default, with exceptions justified on any of the following grounds
Protection of personal data
Protection of commercially confidential information in particular taking into account the MA status of the medicinal product, unless there is an overriding public interest in disclosure
Protecting confidential communication between Member State in relation to the preparation of the assessment report
Ensuing effective supervision of the conduct trial Member States
Where can I find more information?
Information on Clinical trials – Regulation EU No 536/2014, General information, Guideline EU
Ivowen are fully equipped to submit for Clinical Trials Applications on your behalf. Please contact us for more information and for support of your dossier compilation or updates.
Written by: Fiona Downey
https://eureg.ie/wp-content/uploads/2017/05/CT-banner-1.jpeg249299ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2017-05-08 11:38:102023-11-06 11:08:34Clinical Trials Regulation EU No 536/2014 – What does this mean for you?