When will DADI application forms replace the current eAFs?
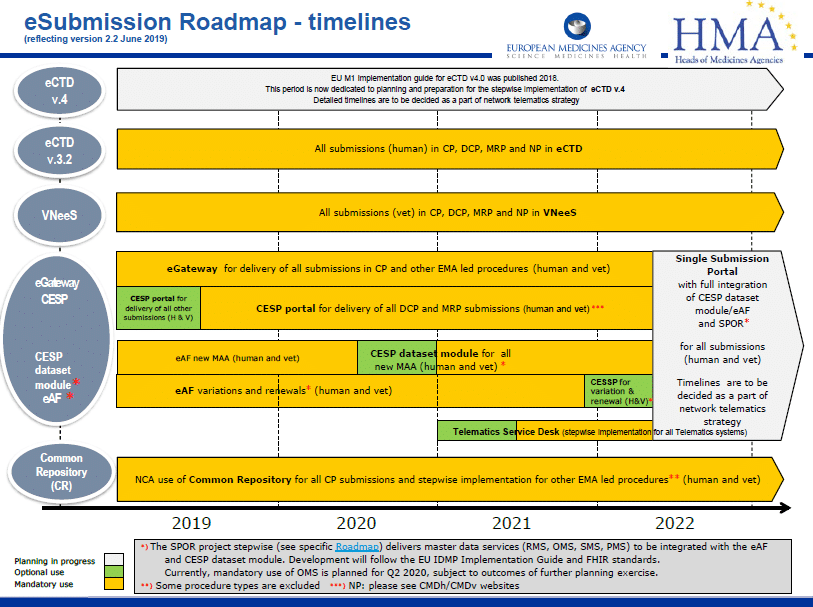
The electronic application forms (eAFs) we are familiar with are in the process of being replaced later this year by a web based digital application form in a new eAF portal. The new eAF portal will look somewhat similar to the current IRIS portal.
This project, known as DADI (Digital Application Dataset Integration), is intended to be used for both CAP (Centrally Authorised Products) & NAP (Nationally Authorised Products) applications to make the future of form-filling and submission-handling more efficient at an EU level.
The Human medicinal product Variation application form will be the first to go live in DADI format. Every person involved in drafting an eAF will need to have an EMA account and user access. Companies who use consultants to prepare eAFs will need to make sure that they assign an EMA role to the consultant.
The next stages of DADI will cover the
- Veterinary variation application form
- Initial MAA form for Human and Veterinary products
- Renewal form for Human and Veterinary products
How will it work?
- The eAF will be filled in using the new eAF portal (via user interface).
- The user will then finalise the eAF by generating a PDF rendition
- This PDF version must still be included in the eCTD submission, as before.
- It will not be possible to submit the form directly from the eAF portal
When will it happen?
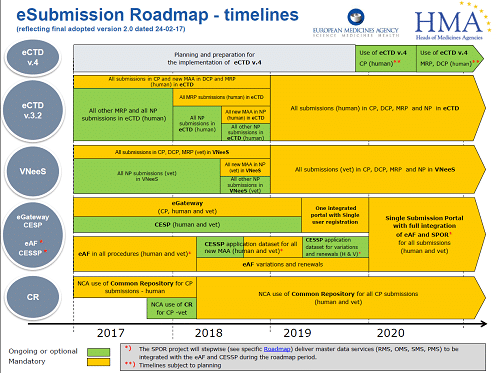
As outlined in the EMA roadmap (link provided below) the two immediate key deliverables are as follows:
| Key deliverables | Go-Live Time Lines | |
| Year | Quarter | |
| Launch of Human variations web-form (parallel use of old and new variation forms as part of a Transition period) | 2022 | Q4 (October) |
| Use of variation web form only | 2023 | Q2 (April) |
It is important for all industry stake-holders to keep up to date with the development of these new web-based forms by consulting the EMA website for updates at the various launch stages.
Where can I find information?
- DADI Network Project Webinar – 18/01/2022 – Live broadcast is available here.
- The updated DADI roadmap, including key milestones, is available here.
- The updated version of the DADI Questions and Answers documents is available here.
- The “Common factors in the Fast Healthcare Interoperability Resources (FHIR) data standard for Art57(2) and eAF”documents are available in the following link.
The project will be implemented in phases, through a set of projects known as SPOR (Substances, Products, Organisations and Referentials) data management services for Human products. The Union Product Database will be the source of data for Veterinary products.
What is SPOR?
| SPOR datasets | Description of data types | Status |
| Substance Management Services (SMS) | Harmonised data and definitions to uniquely identify the ingredients and materials that constitute a medicinal product. | Under Development |
| Product Management Services (PMS) | Harmonised data and definitions to uniquely identify a medicinal product based on regulated information (e.g. marketing authorisation, packaging and medicinal information). | Under Development |
| Organisations Management Services (OMS) | Data comprising organisation name and location address, for organisations such as marketing authorisation holders, sponsors, regulatory authorities and manufacturers. | Operational |
| Referentials Management Services (RMS) | List of terms (controlled vocabularies) to describe attributes of products, e.g. lists of dosage forms, units of measurement and routes of administration. | Operational |
Once the above PMS and SMS are in place, pharmaceutical companies should start preparing to replace their current data submission format in Article 57 Database from the eXtended EudraVigilance Product Report Message (XEVPRM) format to the new ISO IDMP compatible format (HL7 FHIR). Webinars and training will be provided by EMA in due course.
What do Marketing Authorisation Holders have to do at this stage?
- Marketing authorisation holders need to check their data in SPOR (OMS) to ensure it is accurate and up to date. For CAPs the use of OMS data in the current eAF is already mandatory.
- Marketing authorisation holders with authorised MA(s) need to check their data in Article 57 database (xEVMPD) to ensure it is accurate and up to date.
Should you need any support at this stage in getting ready for the new Application Form format please feel free to contact us & the Ivowen team will be here to help.
Written by Marian Winder