2019 – What a year
Ivowen attended the Medicines for Europe conference in January (Regulatory and Pharmacovigilance), the annual EuDRAcon conference in May, exhibited at TOPRA in October and joined our clients from around the world at CPhI in November.
We all saw Brexit come and go, Twice !! We wait to see what lies in store for the next deadline in January 2020.
The FMD came into effect across Europe in February in most member states.
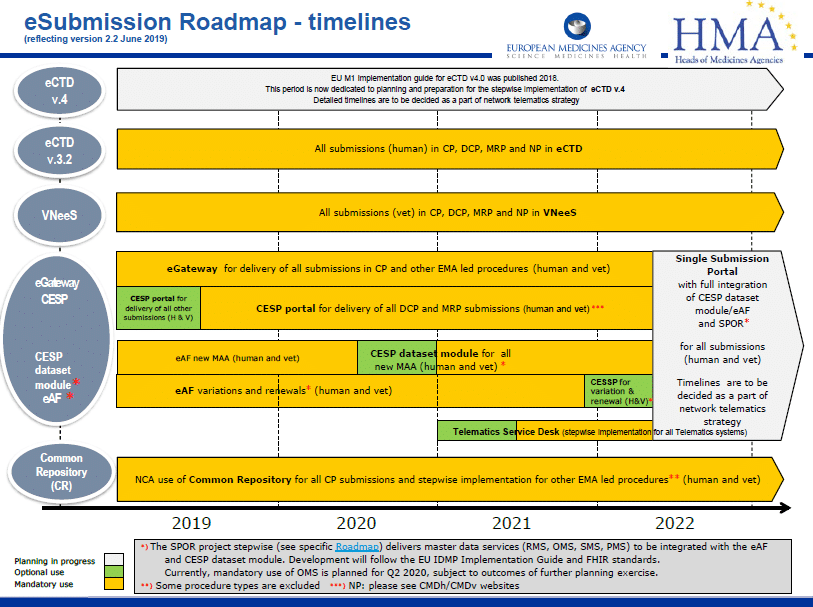
Bulgaria joined CESP, eCTD became mandatory for all human procedures, lots of new guidance was published (to keep us all on our toes) and Nitrosamines in medicinal products moved to the top of everyone’s agenda.
With the festive season now upon us and 2020 on the horizon, Ivowen are setting our sights on the year ahead.
We will be attending the Medicines for Europe conference in January 2020 (Regulatory and Pharmacovigilance) and we encourage you to contact us before mid-January with any specific questions you might like us to ‘ask the regulators’. This is a great opportunity to ask those difficult questions that you just could not get a straight answer to in 2019, on the ever present grey areas of Regulatory procedures.
To help you to plan ahead here are some helpful updates, in brief, as full articles will be posted in 2020:
Falsified Medicines Directive – Where we are now:
- Implemented on 9th Feb 2019 in all MS except Greece, Italy and Belgium
- The European Commission has produced a video to explain more about the safety features.
- The HPRA have extended the use and learn period, initially to Sep 2019 and extended it again to end on a phased basis starting from 31st January 2020.
- The MHRA is also taking a pragmatic, flexible approach to how they enforce the new legal requirements.
Nitrosamines
- Step 1: MAHs should conduct a risk evaluation to identify products at risk of N-nitrosamine formation or (cross-)contamination and report the outcome by 26 March 2020 at the latest.
- EMA guidance can be found at https://www.ema.europa.eu/en/human-regulatory/post-authorisation/referral-procedures/nitrosamine-impurities-overview
- CMDh guidance can be found at https://www.hma.eu/226.html#c6548
Ivowen are here to assist you in 2020 and will continue to provide the top quality service you have come to expect from us.
For more information on Ivowen’s services and how we can help you, contact us.
Written by Alice D’Alton.