EMA eSubmission Gateway – tips & tricks
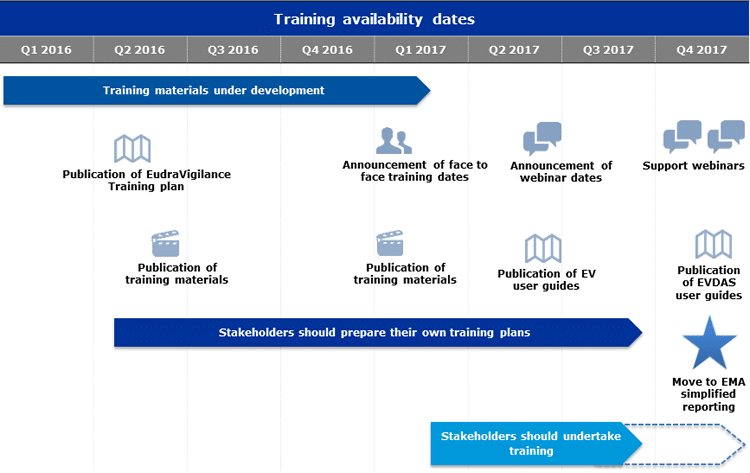
Current State of Play
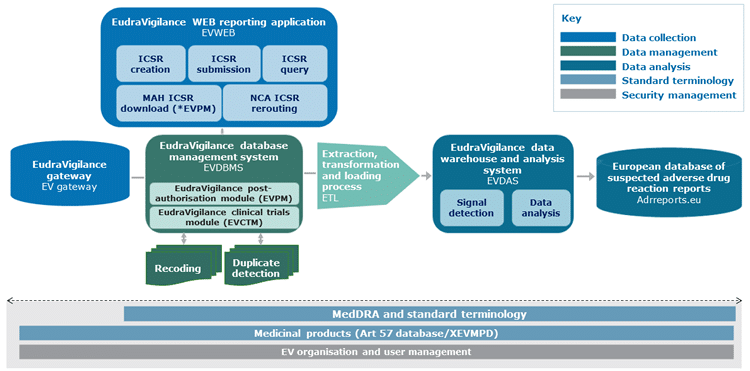
As many of you know, the EMA eSubmission Gateway/Web Client has been mandatory for all submissions for human medicinal products made through the Centralised Procedure since March 2014 and for veterinary products since January 2017. In addition, all PSUSA/PBRER submissions are also made through this EMA eSubmission Gateway, and have been mandatory since June 2016.
Unfortunately, as many of you also know, there are some glitches and issues with this portal. Below are the workarounds for some of these glitches.
Contact us
Please feel free to contact us if you need any help submitting through the EMA eSubmission Gateway/Web Client
“Click to send documents totalling more than 10 MB”
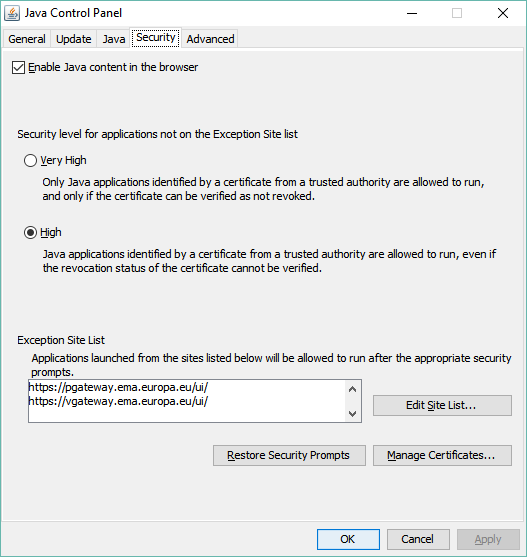
To send any submission through the portal and get an acknowledgement (“ACK”) for this submission, you have to use the “Click to send documents totalling more than 10 MB” option. When you first log in or register for the EMA eSubmission Gateway, and go to the “SEND DOCUMENT” page, this option is not available. This is down to Java settings and security. The way around this is to set your Java security settings and exceptions as follows:
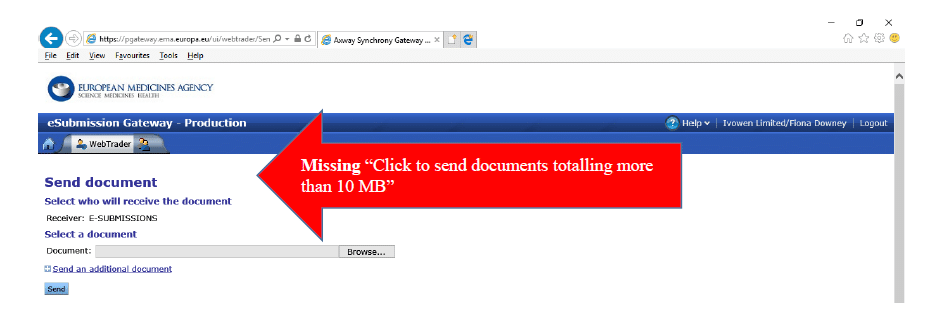
At the moment, you may find that the “Click to send documents totalling more than 10 MB” option is still not available, even when your security settings and exception list are correct. The EMA Service Desk has assured me that they are working on this issue, but in the meantime you can follow the instructions below to work around this issue/
- Open Internet Explorer and log into EMA portal. Go to “Send document”.
- If you see the “Click to send documents totalling more than 10 MB” then continue to send your submission as normal.
- If you don’t see the “Click to send documents…” link, like this example, then follow the instructions below:
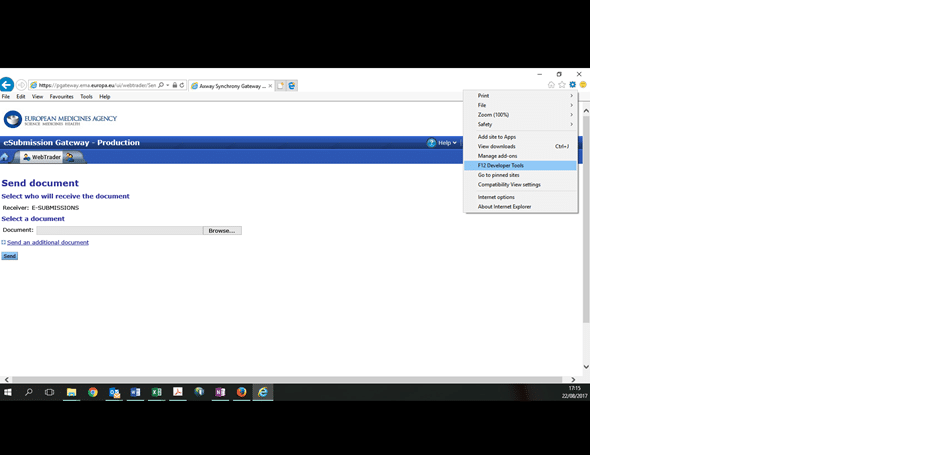
3.1 In the top right hand corner is a Settings button that looks like a machine wheel
3.2 Click on this Settings button and then click “F12 Developer Tools”:
3.3 If you see that the Document Mode is “11 (default)” like the screenshot below:
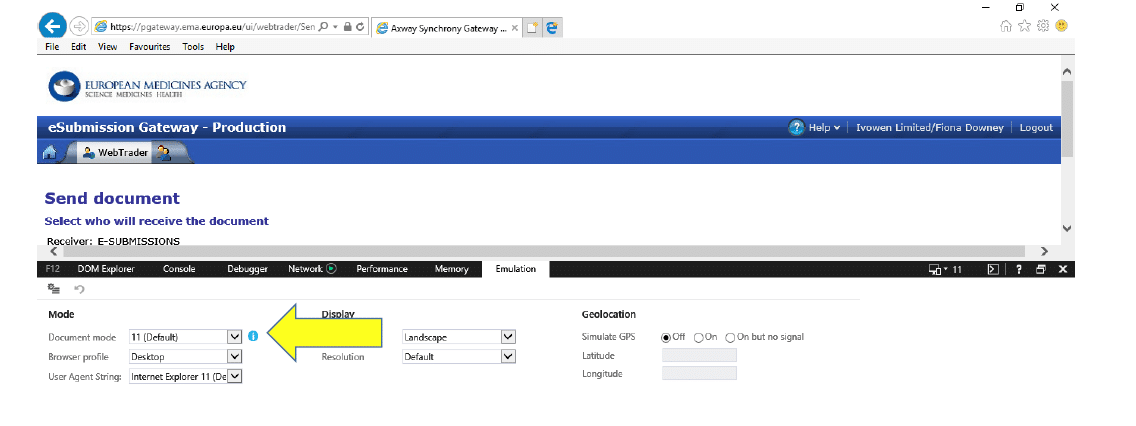
3.4 Change this to “10” and the “Click to send documents…” will appear:
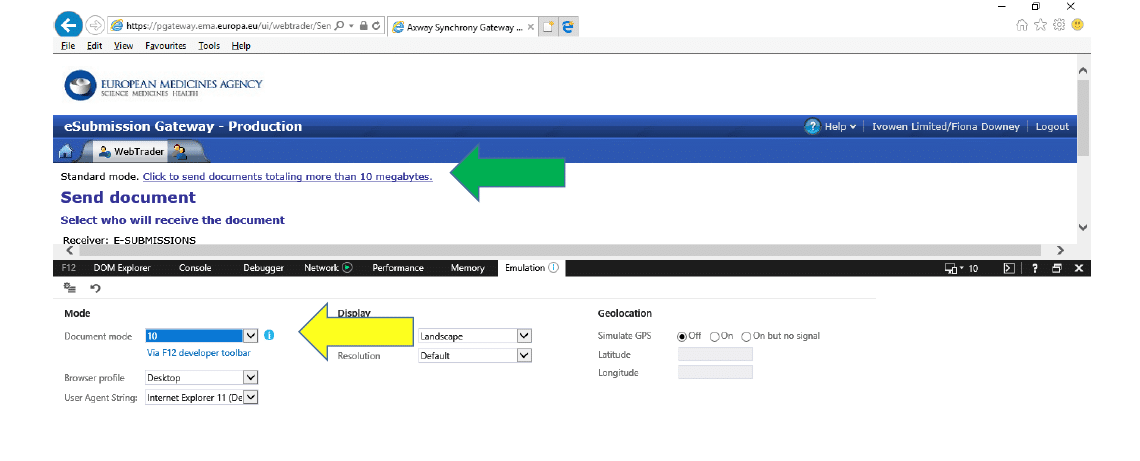
3.5 You need to leave the F12 Developer Tools window with these settings OPEN until you have completed your submission. The EMA Gateway Service desk is working on a fix for this problem, but this is the workaround for now.