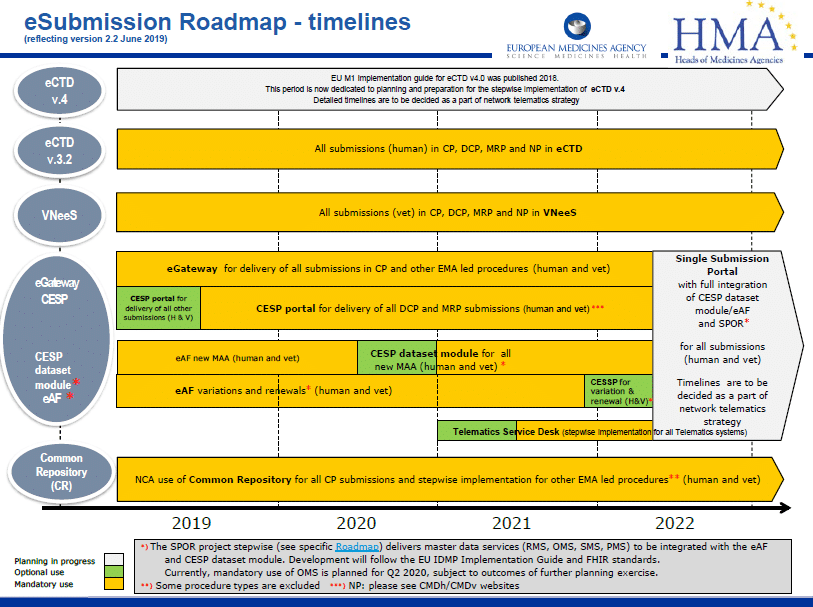
The European Union updated its eSubmission Roadmap in June 2019 to reflect changes in timelines.
What is it?
The eSubmission roadmap is a high level, strategic plan for business and technology changes within the EU. Its function is to align the plans and implementation timelines of target groups and stakeholders, including the EMA, National Competent Authorities (NCAs) and the pharmaceutical industry.
Full details on the eSubmission Roadmap are available on the EMA’s eSubmission website here.
What is happening?
Centralised Procedure(s) (CP)
It is mandatory since January 2010 that all submissions in the CP are made in the eCTD format
National Procedure(s) (MRP & DCP and national only procedures)
It is mandatory since July 2015 and January 2017 that all submissions for new MAAs using the DCP and MRP respectively are made in the eCTD format. For purely national procedures, eCTD has been mandatory for new MAA submissions since July 2018.
For all new submissions in MRP and DCP, the mandatory use of eCTD has been in force since January 2018, and for all new national submissions, since January 2019.
What does this mean?
This means that ALL submissions in the EU for human medicinal products now have to be made in eCTD format. Are you ready? If not, contact us to find out how we can help switch all your current MAs to eCTD and manage all your lifecycle needs. Ivowen has been eCTD compliant since 2009, and therefore has a wealth of experience to become your key partner in this vital step of your registration process.
If you need any clarification or support to manage the changeover to eCTD, Ivowen will gladly work with you to ensure a seamless and efficient transition to eCTD. Contact us for more information or to make an enquiry.
The European Union is arguably the world’s most powerful bloc and very soon it’s about to lose the United Kingdom, one of its biggest members. How and when the UK leaves the EU will have further implications that ripple around the globe.
So if you’ve heard about Brexit but haven’t been keeping up with every twist and turn of the developments, no worries! Ivowen team will provide you with everything you need to know to have your products designed for UK and Brexit affected markets authorised successfully.
What is happening?
EMA
The European Medicines Agency (EMA) will physically relocate to the Netherlands in early March 2019.
EMA will leave its premises in London on 1 March 2019.
It was confirmed that from 4 to 8 March, the Agency will operate on the basis of extended teleworking. During the course of the following week EMA staff will gradually move into the Spark building.
From 4 March 2019 onwards the official address of EMA will be that of the permanent building, located in Amsterdam Zuidas:
European Medicines Agency, Domenico Scarlattilaan 6, 1083 HS Amsterdam, The Netherlands
Meetings and visits will take place at the Spark building:
Orlyplein 24, 1043 DP Amsterdam, The Netherlands
UK guidance on Brexit
Following the outcome of the EU referendum, MHRA still feels responsible for playing a crucial role in medicines and devices regulations as well as vigilance and market surveillance.
As part of the MHRA response to exiting the EU the following Brexit guidance was issued:
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
MHRA guidance on Brexit
Bearing in mind the worst-case scenario if the UK leaves the EU with no deal, the UK would no longer be part of the EU medicines and medical devices regulatory networks and consequently submissions related to human medicines would need to be submitted directly to the MHRA.
The webinar below is relevant for all pharmaceutical companies involved in making medicines regulatory submissions and vigilance activities. It also ensures that stakeholders can be informed of any IT plans and preparations. There is also a section on how all medicines related clinical trial sponsors will register and submit:
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
UK legislation on medicines and medical devices
Legislation has been published which, in the event of the UK leaving the EU with no agreement, will cover the regulation of medicines, medical devices and clinical trials and allow for the continued sale. The Brexit guidance is available here:
The 2012 Regulations (as amended by the 2019 Regulations) make reference to various pieces of EU guidance, as that stood immediately before the exit day (29 March 2019).
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
EU Commission and EMA Q&As
The EU Commission & EMA have published an updated list of questions and answers related to the UKs withdrawal from the EU on the 1st February:
This confirms that dual labelling between UK & Ireland is acceptable where the labels meet the requirements of the Directive and reflect the SPC in Ireland (see Q24).
The focus of this Q&A is on the regulation of medicinal products within the centralised procedure.
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
Brexit Stakeholder Event
Brexit Stakeholder Event – Ivowen was there
Following the UK’s departure from the European Union, the HPRA, together with medicines agencies in Europe, is making preparations to ensure continuity to deliver on patient and animal health remits even if the UK fully exits the current systems as scheduled. There are potential implications for the European network as a whole and particularly for Ireland with its shared marketplace, see meeting agenda below:
Contact us if you would like some more information on this event or Brexit in general
Written by Karolina Dobrychłop
https://eureg.ie/wp-content/uploads/2019/02/Brexit3.jpg437625ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2019-02-19 17:53:522023-11-06 11:15:36Brexit – because it affects you too…
Ivowen have provided some practical guidance and documents below, which may help you prepare for Brexit for products authorised by the DCP and MRP procedures.
Do you still need to change your MAH(s) in any of the EU Member States because of Brexit?
If so, refer to the following updated guidance issued in January 2019 which details the documentation and requirements to submit a Marketing Authorisation Holder transfer:
If you need any clarification or support to complete a Marketing Authorisation Holder transfer, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
Is the UK your current RMS?
If so, you need to initiate an RMS switch as soon as possible. The guidance issued in July 2018 along with the template issued in June 2018 is required to complete this process. This template needs to be completed and sent to a specific e-mail address at the proposed new RMS. The list of contacts points to send the completed template is provided below:
If you need any clarification or support a RMS switch, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
Do you need to change your UK batch release site, etc., if it currently within the UK?
If so, you can refer to the updated guidance document issued in December 2018, which provides information on the type of variation that is required to change functions such as batch release site(s), QC testing site(s), packaging site(s), deletion of site(s) for batch release and changes to the QP for Pharmacovigilance (QPPV) and PSMF where these functions still reside within the UK:
If you need any clarification or support to complete variations to support changes needed as a result of Brexit, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
Do you supply product to Cyprus?
If so, the Cypriot Competent Authority has issued a newsletter to Stakeholders on the 14 January 2019 which lists 105 products authorised via an exceptional marketing authorisation affected by Brexit which are considered critical. The Drugs Council in Cyprus recommends that pharmaceutical companies register these products via an exceptional marketing authorisation using another reference state other that the UK.
If you need any clarification or support to complete an Exceptional Marketing Authorisation to avoid no supply of these critical products on the Cypriot market, Ivowen will gladly assist you in a timely manner. Contact us for more information or to make an enquiry.
As the date for the withdrawal of the UK (also known as Brexit) from the EU approached closer, it pays to hope for the best and prepare for the worst. With this in mind, the EMA have issued a guidance document on “Practical guidance for procedures related to Brexit for medicinal products for human and veterinary use within the framework of the centralised procedure, EMA, 19 June 2018” to prepare for the UK’s exit from the EU by the 30 March 2019.
For marketing authorisation applications (MAAs) that are expected to receive a Commission Decision (i.e. be approved) after 29 March 2019, the QPPV, PSMF (for medicines for human use), batch release sites, batch control sites, intended OMCL (if applicable) and nominated local representatives for Member States other than UK must be located in in the Union (EEA).
As published on the EMA website on the 10 July, a recent European Medicines Agency (EMA) survey shows that marketing authorisation holders for more than half (58%) of the 694 centrally authorised products (CAPs) with an important step in their regulatory processes in the United Kingdom (UK), are on track with their regulatory planning to ensure that their marketing authorisation remains valid once the UK leaves the European Union (EU).
This also means that 42% are NOT ready… Are you one of the 42%
The EMA urges those companies who have not yet informed EMA of their Brexit preparedness plans to do so as soon as possible to mitigate any risks to the continuous supply of medicines for human and veterinary use within the EU.
We can help…
Ivowen can help Clients prepare to implement the required changes by providing the following services:
Provide practical guidance on what needs to be in place by the above date to address situations where the UK is the current MAH, batch release site, batch control sites, location of QPPV& PSMF, transfer of orphan designation, etc.
Act as a MAH in the EU
Provide Pharmacovigilance (QPPV) services in the EU
Provide Pharmacovigilance System Master File and backup services located in the EU
Assist Clients in selecting and transferring RMS to another EU member state where the UK is current MAH
Preparation of Brexit related variations to be ready for above timelines
Assistance in changes needed to Product Information to reflect changes such as new MAH, batch release site(s), amend names of local representatives
Please contact us for further information at any time.
https://eureg.ie/wp-content/uploads/2018/07/Brexit.jpg351624ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2018-07-31 15:09:352023-11-06 11:15:27Preparing for Brexit
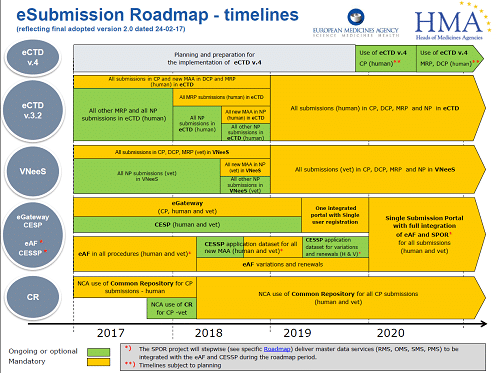
All good (and bad) things, must come to an end. The NeeS (Non-eCTD electronic submissions) submission format, will no longer be accepted for any Marketing Authorisation applications (MAAs) or submissions in MRP or DCP from 1st January 2018. Centralised Procedure (CP) submissions have been mandatory in eCTD since 2010. All new MAAs submitted using either the Decentralised Procedure (DCP) or the Mutual Recognition Procedure (MRP), have had to be in eCTD format since the middle of 2015. As of 1st January 2018, all MRP submissions must be submitted in eCTD. Therefore, if your application/product is not in eCTD format and you need to submit an MRP variation, you have to transition to eCTD for the next submission.
What is the timetable?
Starting from 1st January 2018 it is mandatory for all CP, DCP and MRP submissions to be in eCTD format. For national procedures (NP) the deadline is currently set at 1st July 2018 for new MAAs.
Following quickly behind these requirements, all submissions in CP, DCP, MRP and NP will have to be in eCTD from January 2019. At this stage, the exact date is not confirmed, but you need to be ready.
We can help…
Ivowen can create a baseline dossier for any submission based on currently approved information. Currently, baselines are not mandatory but some Member States are requesting them. In addition, it is best practise to get your dossier into a baseline for all future submissions.
Ivowen can transition your application to eCTD at the next regulatory activity (variation, renewal, Article 61.3, etc.).
Ivowen can manage the eCTD lifecycle for you in-house and provide you with fully valid, submission-ready sequences.
Contact us for more information or for help with building your eCTD now.
https://eureg.ie/wp-content/uploads/2018/02/2018-02-eSubmission-Roadmap-final-slide-v2.0-Feb-2017..png373500ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2018-02-07 12:22:002023-11-06 11:12:36eCTD is coming…
As many of you know, the EMA eSubmission Gateway/Web Client has been mandatory for all submissions for human medicinal products made through the Centralised Procedure since March 2014 and for veterinary products since January 2017. In addition, all PSUSA/PBRER submissions are also made through this EMA eSubmission Gateway, and have been mandatory since June 2016.
Unfortunately, as many of you also know, there are some glitches and issues with this portal. Below are the workarounds for some of these glitches.
Contact us
Please feel free to contact us if you need any help submitting through the EMA eSubmission Gateway/Web Client
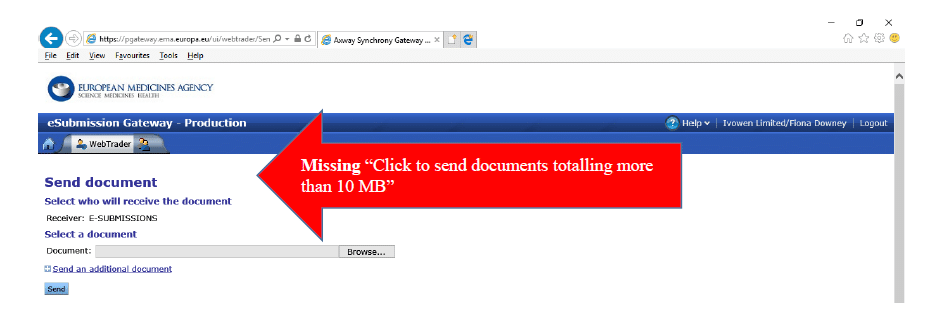
“Click to send documents totalling more than 10 MB”
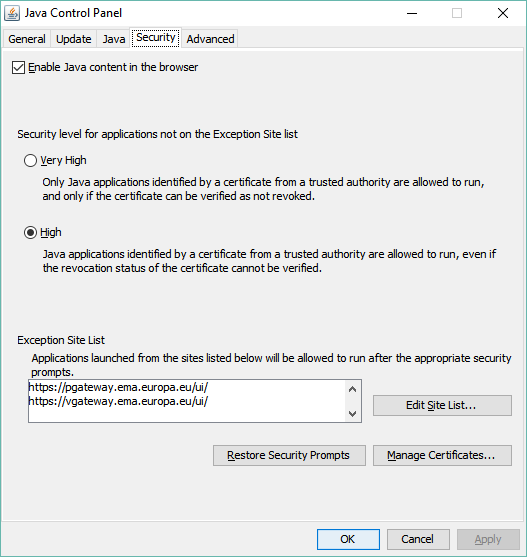
To send any submission through the portal and get an acknowledgement (“ACK”) for this submission, you have to use the “Click to send documents totalling more than 10 MB” option. When you first log in or register for the EMA eSubmission Gateway, and go to the “SEND DOCUMENT” page, this option is not available. This is down to Java settings and security. The way around this is to set your Java security settings and exceptions as follows:
At the moment, you may find that the “Click to send documents totalling more than 10 MB” option is still not available, even when your security settings and exception list are correct. The EMA Service Desk has assured me that they are working on this issue, but in the meantime you can follow the instructions below to work around this issue/
Open Internet Explorer and log into EMA portal. Go to “Send document”.
If you see the “Click to send documents totalling more than 10 MB” then continue to send your submission as normal.
If you don’t see the “Click to send documents…” link, like this example, then follow the instructions below:
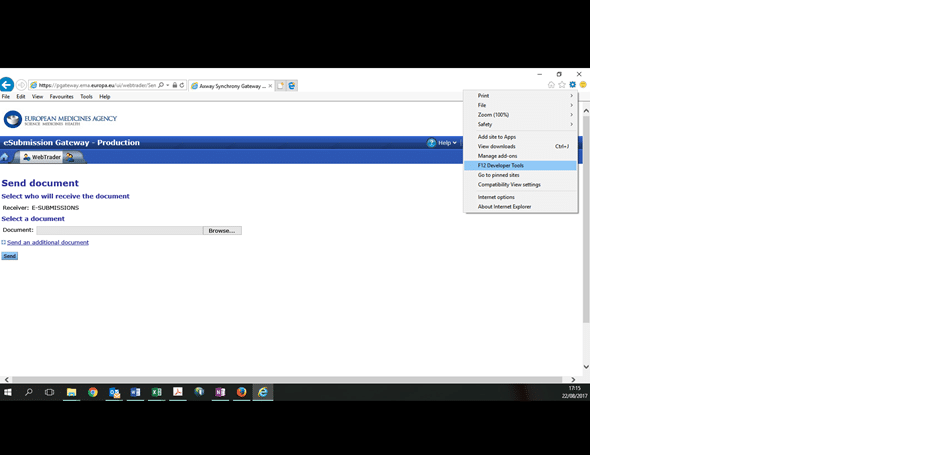
3.1 In the top right hand corner is a Settings button that looks like a machine wheel
3.2 Click on this Settings button and then click “F12 Developer Tools”:
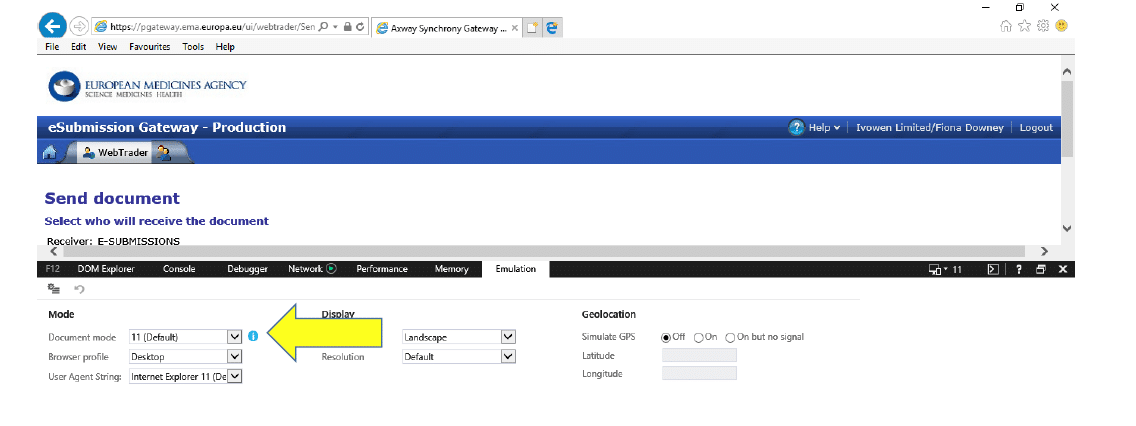
3.3 If you see that the Document Mode is “11 (default)” like the screenshot below:
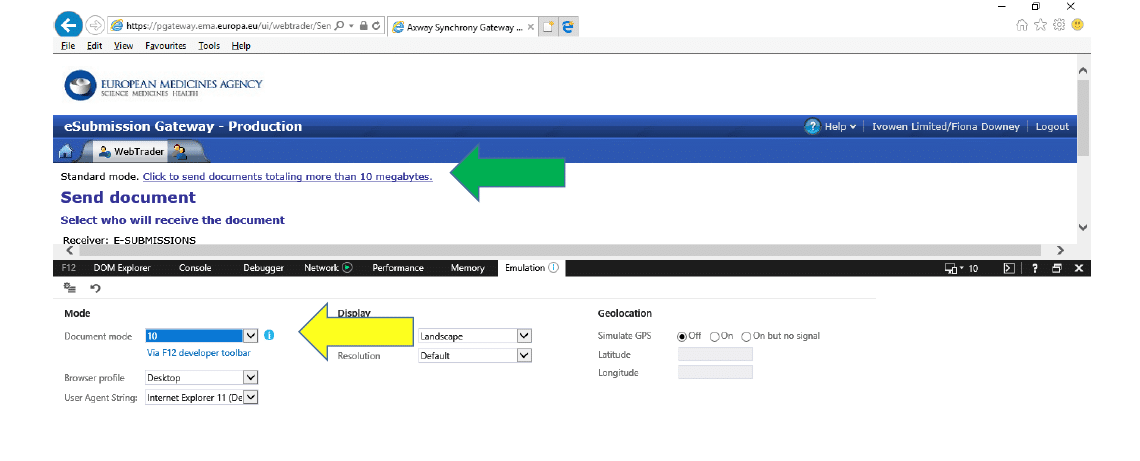
3.4 Change this to “10” and the “Click to send documents…” will appear:
3.5 You need to leave the F12 Developer Tools window with these settings OPEN until you have completed your submission. The EMA Gateway Service desk is working on a fix for this problem, but this is the workaround for now.
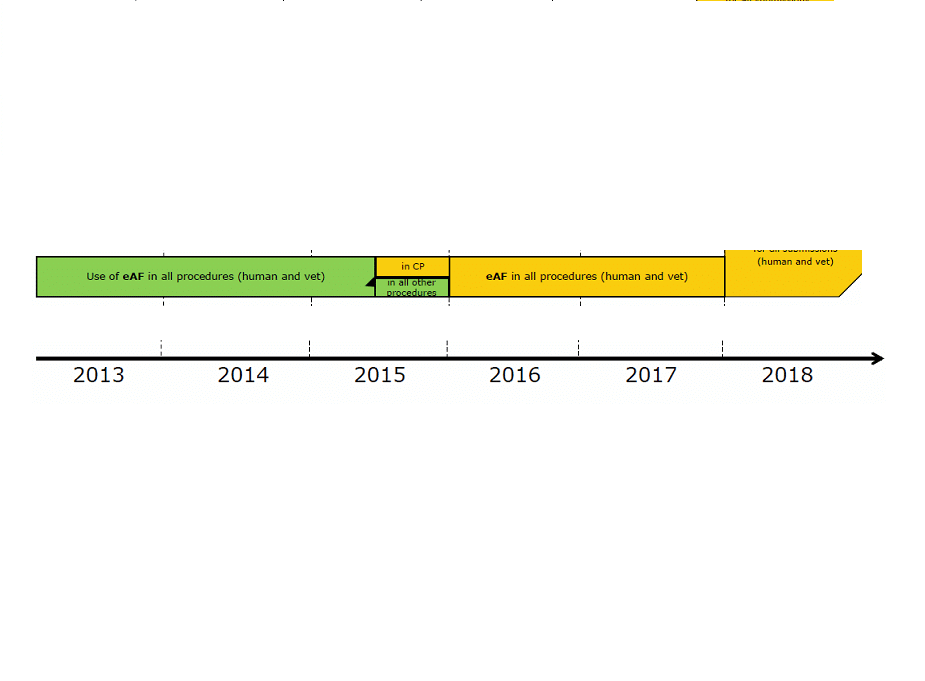
The eAF is mandatory for all procedures from 01/01/2016 (CP, MR, DC, National in eCTD/NeeS/Paper). However, Presubmission Meeting Requests, Article 61(3) & MAT forms remain the outside scope and can be submitted as paper or PDFs.
Please contact us if you require any assistance with any of the forms or any advice on any of the above procedures. For your convenience, the following is a summary of the important aspects of using the eAF.
Word versions will be removed from Notice to Applicants in January 2016
After 11/01/2016 only version 1.19 will be acceptable for new procedures
eAF should be printed for Paper submissions
Version 1.20 is planned for April 2016 (unless hotfixes are required before then)
It is important to note, and welcome news, that there is no need to update to a newer version of the eAF in the middle of a procedure
See the guidelines (update planned in January 2016) and release notes (lists the changes made to newer versions) for further information
In line with the proposed move towards a Single Submission Portal (SSP) in place of CESP and the EMA Gateway, there are plans to reformat the eAF into a data capture system that can be submitted directly through CESP. This is in a very preliminary stage.
Technical queries should be sent to EMA Service Deskvia IT service portal (login)
Q&A documents should be consulted first
Fast web view warning in the dossier validation report for the eAF can be ignored
Procedural queries should be directed to the National Competent Authority (NCA) (and response sent to EMA Service Desk). Complicated queries should be sent to both parties.
Webinars are available for the NCAs (to try to reduce the requests for MS specific national requirements)
Using the eAF
Technical Validation of the form (internal):
Once signature is added and form validated, it is now locked. Locked forms cannot be amended. Therefore, the signature should be the last thing added to the form.
Always keep a copy of the unlocked form so that amendments can be made (e.g. during preparation or for requests from NCA for updated forms during a procedure)
Do not use bookmarks as these may cause invalidation issues
Annexes to the form should be filed separately in module 1.2 (do not use the PDF function to insert them into the eAF)
Form should be named; cc-form-eaf-var
Annex should be named; cc-form-annex-var (e.g. cc-form-5-19 or cc-form-proofpayment)
Electronic signature can be an image of a real signature (e.g. jpeg file – a scanned copy of wet signature. This however is not an electronic signature and is only used to close/lock the form) or can also be a line of text which states that signature is on file internally (e.g. “This form was authorised following company policies by Majella Ryan, Regulatory Affairs Manager of Ivowen with authorisation to sign. The signature is on file”)
The eAF does not accommodate multiple signatures at present. A separate annex should be provided if multiple signatures are mandatory for a particular NCA. Multiple name sections will be incorporated into version 2.0 but no mention of whether multiple signatures will also be accommodated.
The signature should be provided by the responsible MAH or can be provided by any authorised deputy
Please also check national requirements for signatures.
See Q&A guidance for further information
Workaround solutions (e.g. Annex B for multiple MAH or Product names) should always be mentioned in your cover letter
Some unforeseen variations (category z) are not adequately accounted for yet. Details of such changes should be outlined in the scope section of form and in the cover letter.
Duplicate sections only if products differ with regard to API or Pharmaceutical Form
Annex A or Annex B can be used for multiple MAH or Product names. Click on Annex A/B button in form and add the annex as a separate file in Module 1.2
Detailed instructions on how to use workaround solutions is available in the eAF Q&A document and the eAF Technical Guidance documents – both are published on the eAF webpage – if you need more advice contact EMA Service Desk.
No translations of the eAF are available, nor should they be requested.
Drug substances can be entered from controlled vocabulary lists or free text, and these each have different EV codes.
The focus should be on using substance, product, organisation and referential (SPOR)
Strikethrough text function is not available in eAF but text can be copied and pasted (with text struck through) from Word or Outlook
In MRP/DCP one common application form is highly recommended, one per pharmaceutical form or strength for all member states in case of new MAA and one eAF for all involved products for all member states in case of variations and renewals.
Written by Majella Ryan
https://eureg.ie/wp-content/uploads/2016/02/eSub-roadmap-eAF-02-2016-1.png681925ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2016-02-03 16:03:552023-11-06 11:08:23Points to note on the eAF