Back to Basics – Regulatory 101 – Legal basis of your MA application in the EU and UK
With our new logo and new website we expect some new visitors, so, I decided to go back to basics for those who may be new to Regulatory Affairs.
(If you need help with the jargon visit our A-Z Glossary of Regulatory abbreviations)
So, let’s start with the Legal Basis which forms the first step of your MA application (MAA for short). You need to know the legal basis before you can finish compiling your application.
The legal requirements and necessary procedures for submitting an initial application for an EU marketing authorisation (MAA) are set out in Directive 2001/83/EC, as amended, and in Regulation (EEC) No. 726/2004 (the latter for CP applications).
The legal requirements for a UK MAA are set out in the Human Medicines Regulations.
The relevant articles and regulations from this legislation are provided below followed by a brief description of each legal basis:
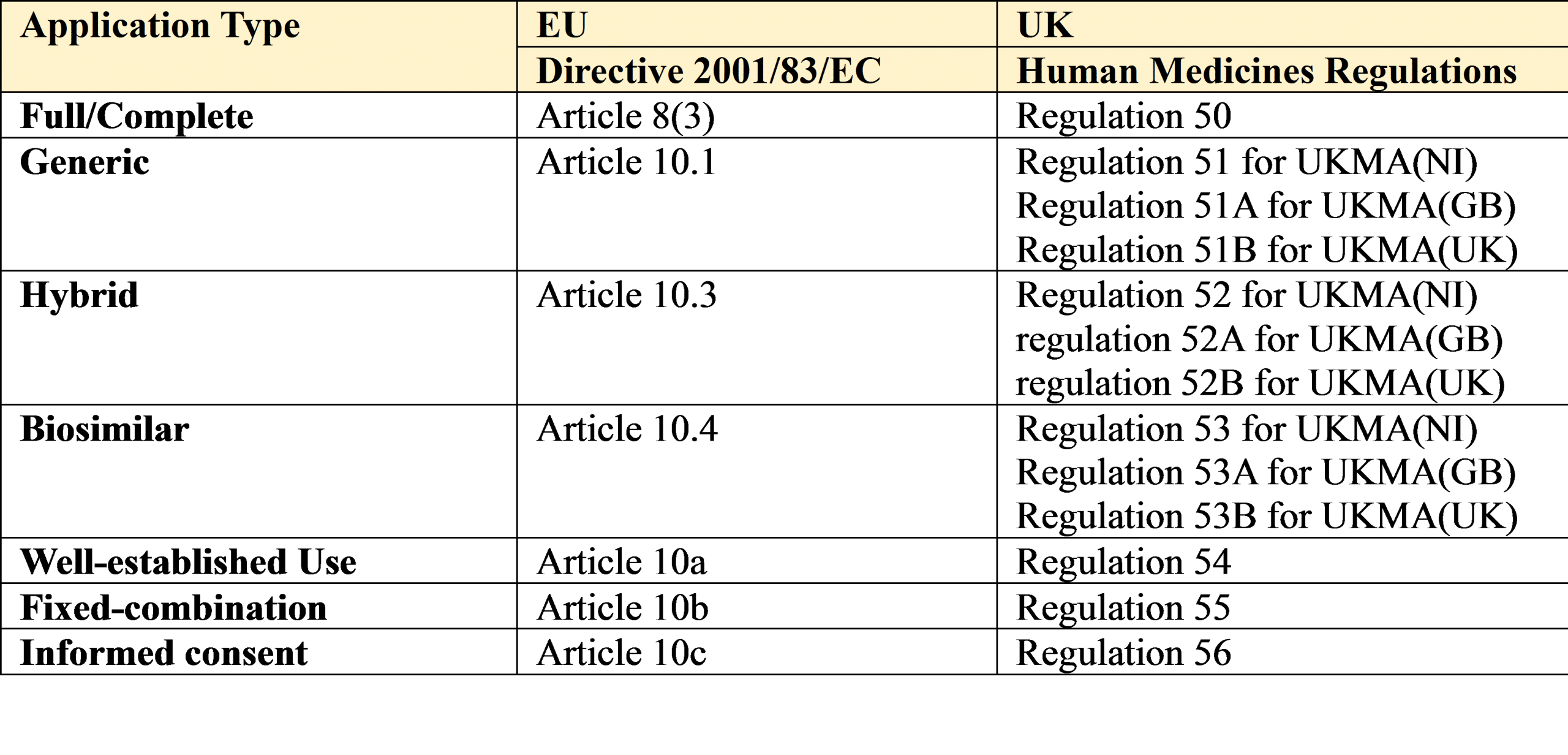
1. Full/Complete
An application for a marketing authorisation (MAA) must be accompanied by the particulars set out in Article 8(3) of Directive 2001/83/EC, as amended, and as well as all the usual documentation will also require the results of
• Physico-chemical, biological or microbiological tests,
• Pharmacological and toxicological tests,
• Full Clinical trials in human subjects
This Legal basis is most often used for brand new medicines (Drug Product) or active substances (Drug Substance) which have never been authorised in EU or UK before. It results in 10 years or 8+2+1 years of data exclusivity. No generic version can be marked until this exclusivity expires.
2. Generic
This is used when the medicinal product (Drug Product) for which an MA is sought is considered essentially similar to and interchangeable with a medicinal product that has already been authorised via Article 8(3) in the EU or UK.
Essentially similar means it has the same active substance, the same amount of active substance (strength), and the same pharmaceutical form.
In this case the Applicant cross refers to an already approved EU/UK reference medicinal product (RMP for short) to use its already approved data on the Pharmaco-toxicological and Clinical aspects of the application and so does not need to repeat these trials. Instead a bioequivalence study in human subjects is required. This data will show that the product is safe and works the same way inside the human body as the RMP does.
3. Hybrid
These are applications which rely on a mixture of data – i.e. a cross reference to the RMP for some aspects of the clinical and pre-clinical data requirement plus provision of additional data in support of any aspect of the proposed drug product which is different to the RMP (i.e. where it is not essentially similar).
This type of application is used when the proposed product has a different/new indication, different/new dosage form, different/new strength, etc. that is not currently approved for the RMP.
4. Biosimilar
These are generic products which are derived from a biological source rather than a chemically synthesised molecule.
It is often difficult to treat biosimilar generic products in the same way as we treat ‘regular’ generics as it is very tricky to prove essential similarity in the conventional ways – therefore provision of some preclinical and clinical data may be necessary to prove safety and efficacy of the biosimilar product.
The Guideline on similar biological medicinal products should be consulted when considering using a non-EEA authorised comparator (i.e. a non- EEA authorised version of the RMP) in the development of a biosimilar.
5. Well Established Use/Bibliographical
It is possible to replace results of pharmacological and toxicological tests or clinical trials with detailed references to published scientific literature if it can be shown that the drug has a ‘well established use’ with recognised efficacy and known safety.
The minimum criterion for using this legal basis of application is that the drug substance concerned has to have been marketed (i.e. in use) in the EU for at least 10 years. It is commonly used when no RMP exists to refer to or to perform a bioequivalence study against.
This type of application, although allowed for under the legislation, is not always accepted by member states (and they have been known to request clinical data, during assessment) but with the use of real-world data and evidence on the increase this type of application may become more common. Only Time will tell.
6. Fixed Combination
In the case of new drug products containing known active ingredients (drug substances), not before used in combination for therapeutic purposes, the results of pharmacological and toxicological tests as well as clinical trials in human subjects, relating to that combination, must be provided.
This legal basis covers entirely new products that are a mixture of two or more drug substances (sometimes called monocomponents), which have already been authorised and marketed in the EU in their own right. Because this is theoretically a full, stand-alone application (not unlike Article 8(3)), the product enjoys the full 10 or 8+2+1 years data of exclusivity.
Please note that the concept of the global marketing authorisation applies here so once a combination is approved any new application(s) for the ‘same’ product cannot be considered new combinations anymore and so an article 10b would not be acceptable in this situation.
7. Informed consent/Duplicate
The Marketing Authorisation Holder (MAH for short) of a complete/standalone dossier (i.e. the originator/innovator/RMP) can consent in writing to allow a new applicant/MAH to refer to the contents of their approved MA (on file with Competent Authority) for the purpose of obtaining a new MA.
The reference dossier must be a complete dossier (Article 8(3)) and consent must be obtained for all three modules containing the pharmaceutical (Module 3), preclinical (Module 4) and clinical data (Module 5).
The Applicant must choose the correct legal basis in advance of submission and selecting the wrong one will delay your application.
If you need any clarification or support contact us and we will gladly assist you.
If you are starting out in Regulatory Affairs and you now have a long list of questions after reading this article, we can help. We provide detailed training for all levels of Regulatory experience. You or your manager can contact us for details and our training menu.
Written by

Fiona Downey 1