The submissions may concern national and European procedures (MRP, DCP) both for human and veterinary medicinal products.
Also from 3rd April 2017 the following submissions should be transmitted through CESP for Greece:
All submissions for Mutual Recognition/Decentralised Procedures (MRP/DCP)
national procedures relating to approvals, variations, renewals, PSURs, ASMFs etc., as well as any other documentation should be submitted using CESP.
Responses to deficiencies and any other additional information that can be requested from the EOF within the claims of assessment.
What does this mean for you?
To date in Poland, only submissions concerning initial MAAs were possible via CESP. There are still some national requirements in Poland, however. All documents that require an original signature can be either submitted
in electronic version with a valid electronic signature or
must be submitted in parallel to CESP in paper.
Documentation once submitted via CESP should be submitted via this channel throughout its whole lifecycle.
For submissions into Greece, additional CD/DVDs are no longer required. In addition, for each submission, a corresponding reference number will be automatically assigned and will be sent directly to the applicant.
Where can I find more information?
Full details are available on the Polish Ministry’s website (in Polish) here.
Ivowen are fully equipped to prepare and submit any applications on your behalf. Please contact us or our EuDRAcon partners for more information and for support of your dossier compilation or updates.
Written by Majella Ryan
https://eureg.ie/wp-content/uploads/2017/04/CESP-front-page.png4951477ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2017-04-05 15:48:172023-11-06 11:08:23CESP expands in Poland and Greece
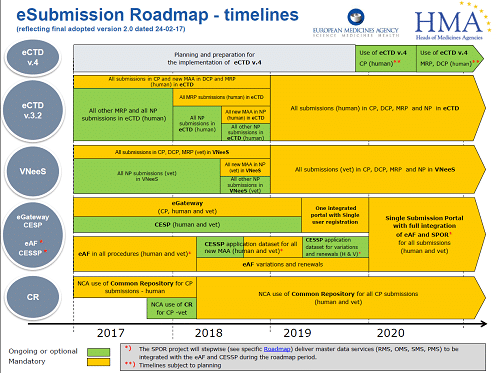
All good (and bad) things, must come to an end. The NeeS (Non-eCTD electronic submissions) submission format, will no longer be accepted for any Marketing Authorisation applications (MAAs) or submissions in MRP or DCP from 1st January 2018. Centralised Procedure (CP) submissions have been mandatory in eCTD since 2010. All new MAAs submitted using either the Decentralised Procedure (DCP) or the Mutual Recognition Procedure (MRP), have had to be in eCTD format since the middle of 2015. As of 1st January 2018, all MRP submissions must be submitted in eCTD. Therefore, if your application/product is not in eCTD format and you need to submit an MRP variation, you have to transition to eCTD for the next submission.
What is the timetable?
Starting from 1st January 2018 it is mandatory for all CP, DCP and MRP submissions to be in eCTD format. For national procedures (NP) the deadline is currently set at 1st July 2018 for new MAAs.
Following quickly behind these requirements, all submissions in CP, DCP, MRP and NP will have to be in eCTD from January 2019. At this stage, the exact date is not confirmed, but you need to be ready.
We can help…
Ivowen can create a baseline dossier for any submission based on currently approved information. Currently, baselines are not mandatory but some Member States are requesting them. In addition, it is best practise to get your dossier into a baseline for all future submissions.
Ivowen can transition your application to eCTD at the next regulatory activity (variation, renewal, Article 61.3, etc.).
Ivowen can manage the eCTD lifecycle for you in-house and provide you with fully valid, submission-ready sequences.
Contact us for more information or for help with building your eCTD now.
https://eureg.ie/wp-content/uploads/2018/02/2018-02-eSubmission-Roadmap-final-slide-v2.0-Feb-2017..png373500ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2018-02-07 12:22:002023-11-06 11:12:36eCTD is coming…
Falsified medicines are fake medicines that are designed to mimic real medicines. Due to the increase of falsified medicines on the market the EU has a strong legal framework for licensing, manufacturing and distribution of medicines. In July 2011 DIRECTIVE 2011/62/EU (http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2011:174:0074:0087:EN:PDF ) came into force; this directive aims to prevent falsified medicines entering the legal supply chain and thus reaching patients. One of the Directive’s measures is the introduction of safety features on medicines.
Update on Safety Features on Medicines:
On February 9th 2016 the EC published an implementation plan for the introduction of the safety features on the packaging of nationally authorised medicinal products for human use:
In conjunction, a Regulation was also published laying down detailed rules for the safety features appearing on the packaging of medicinal products for human use:
The Delegated Regulation will apply in all European countries from the 9th February 2019 (3 years after its publication). Belgium, Greece and Italy have the option of deferring the application of the rules by an additional period of up to 6 years.
There are two safety features to be placed on the packaging of most prescription medicines and certain non-prescription medicines no later than 9 February 2019.
-1). a unique identifier (a 2-dimension barcode) and
-2). an anti-tampering device (ATD).
How does this affect your medicinal products and applications?
NewMAAs submitted from April 2016:
QRD:
Revised QRD template.
Revised dossier sections:
In the case of medicinal products where the ATD is placed on the immediate packaging because there is no outer packaging and the ATD affects the container and its closure system(s), applicants are required to include information on the ATD and how the ATD affects the container and its closure system(s) (sections 3.2.P.2.4 and/or 3.2.P.7 of the Notice to Applicants Volume 2B)
OngoingMAAs
QRD:
CHMP opinion in March 2016 advised to comply Revised QRD template
CHMP opinion from April 2016 onwards, applicants must comply with the revised QRD template
Revised dossier sections:
As per new MAAs
Existing MAs
QRD:
Revised QRD template within 3 years – Can be implemented in Type IA, Type IB, Type II, Renewals, Line extension etc. where the submission affects the product information (PI). Approval of submissions must be no later than the 9th February 2019. If no regulatory procedure occurs within the required timeframe a notification is requested to be submitted pursuant to article 61(3).
Revised dossier sections:
If the ATD is placed on the immediate packaging and the ATD affects the container and its closure system(s), applicants are required to submit the appropriate variations to include the information on the ATD and how the ATD affects the container and its closure system(s) (see section B.II.e of the Variation Guidelines).
Medicinal product no longer needs to bear safety features
QRD:
Regulatory procedure to remove the standard statements regarding the unique identifier and ATD.
Revised dossier sections:
ATD on immediate packaging: Regulatory procedure to remove the statements regarding the ATD in the dossier. ATD on outer packaging – no regulatory procedure necessary.
Change of Legal Status
QRD:
Non-prescription to prescription following a MAH switch application: MAH should use the regulatory procedure to comply with the revised QRD and regulations. Non-prescription to prescription following a Commission referral or a PSUR assessment, the Commission Decision will cover, inter alia, the regulatory requirements to implement the safety features.
Revised dossier sections:
Non-prescription to prescription: MAH should use the regulatory procedure to include the information on the ATD and how the ATD affects the container and its closure system(s)
What does the new QRD template now include?
The new QRD template includes the following sections in: Particulars to appear on <the outer packaging> <and> <the Immediate packaging:
UNIQUE IDENTIFIER – 2D BARCODE
UNIQUE IDENTIFIER – HUMAN READABLE DATA
Updated QRD template in track changes is available here:
Are any medicinal products not subject to prescription but that should include the safety features above?
Yes, the medicinal products not subject to prescription that shall bear the safety features, referred to in Article 45(2) are listed in Annex II of the Regulation located here:
If you have any queries on the above, if you would like any help with complying with the new regulation or if you have any other queries please contact us .
Written by Emily Fletcher.
https://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svg00ERAadministratorhttps://eureg.ie/wp-content/uploads/2023/10/European-Regulatory-Affairs-Logo.svgERAadministrator2016-03-14 12:47:332023-11-06 11:08:27Falsified Medical Directive (FMD) and updated QRD template released February 2016