
The new veterinary regulation (Regulation 2019/6) and its implications on regulatory submissions for veterinary medicinal products
The new veterinary regulation (NVR), Regulation 2019/6 applied to all EU Member States from 28 January 2022. The new legislation represents a significant change in how veterinary medicinal products are authorised, monitored and controlled in the EU.
The Regulation was developed in order to implement a fit-for-purpose veterinary legislation which would no longer be based on the equivalent human medicines authorisation system.
The legislation repeals Directive 2001/82/EC.
The changes are intended to:
- reduce the administrative burden on companies and regulatory authorities
- enhance the availability of veterinary medicinal products
- stimulate innovation of new and existing medicines
- strengthen the EU response to fight antimicrobial resistance.
The new Regulation 2019/6 is broken down into the following chapters:
I. Subject matter, scope and definitions
II. Marketing authorisations
III. Procedures for marketing authorisations
IV. Post marketing authorisation measures
V. Homeopathic veterinary medicinal products
VI. Manufacturing, import and export
VII. Supply and use
VIII. Inspections and controls
IX. Restriction sand penalties
X. Regulatory network
XII. Common and procedural provisions
XII. Transitional and final provisions
Here is a summary of some of the noteworthy regulatory changes that have been introduced in chapters II, III & IV of the new veterinary regulation:
Chapter II – Marketing Authorisations:
An MA for a veterinary medicinal product shall be valid for an unlimited period of time. Hence, there is no longer a requirement for a renewal procedure or the sunset clause.
Chapter III – Procedures for marketing authorisations
- Decentralised Procedure:
- Scope and timelines remain unchanged
- Responsibilities of RMS, CMS and applicant have changed at some steps of the procedure. For example,
– CMSs will also provide comments directly to the applicant at Day 100 and Day 145 instead of to the RMS only (therefore, comments are no longer anonymised).
– at Day 100-105 and Day 145-150, the applicant will now compile and circulate the LoQs.
– at Day 210, RMS will now be required to circulate a Final Assessment Report (FAR). - Possibility now for re-examination request by applicant according to Article 50 of the NVR. For further information on this change, note that CMDv have published a Best Practice Guide for Re-examination of RMS assessment report procedure.
- Mutal Recognition Procedure:
- Scope remains the same. However, a minimum of six months is required between the decision granting the national MA and submission of an application for a MRP.
- 90 day procedure length remains unchanged but there are changes to some of the time-points in the procedure.
- Responsibilities of RMS, CMS and applicant have changed at some steps of the procedure, similar to those outlined above for the DCP.
- Centralised Procedure:
- Scope of the mandatory use of the procedure has been widened. Refer to Article 42 (point no. 4) for details.
- National Procedure:
- No significant changes.
- Subsequent Recognition Procedure (SRP):
- Previously known as the “Repeat Use Procedure” is now officially recognised under Article 53 of the NVR.
- Timelines and other requirements have been changed.
Due to the changes caused by the new regulation, CMDv have published updated guidance for DCP, MRP and SRP procedures:
Chapter IV: Post marketing authorisation measures (Variations)
In terms of variations to marketing authorisations, one of the main changes arising from Regulation EU 2019/6 is that instead of the previous categories of Type IA, IB and II variations there will now only be two categories of variations:
- Variations Not Requiring Assessment = VNRA
- Variations Requiring Assessment = VRA
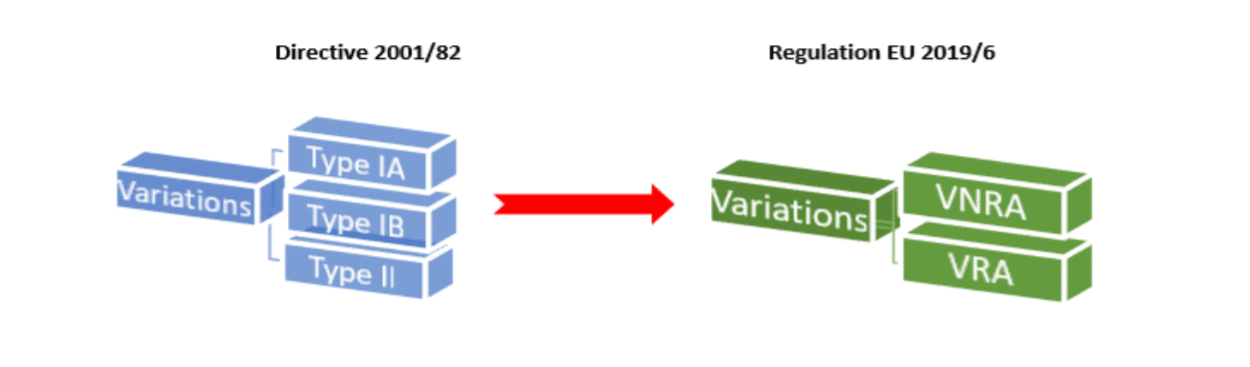
VNRAs consist of all the previous type IA and some Type IB variation categories.
VRAs will consist of most of the previous Type IB and all of the Type II variation categories.
Commission Regulation 1234/2008 will now no longer apply to veterinary medicinal products due to the introduction of new veterinary regulation (2019/6).
Variations Not Requiring Assessment (VNRA)
The Implementing Regulation (EU) 2021/17*, includes a list of all variations not requiring assessment along with any associated conditions and documentation requirements and is published in the EU Official Journal here.
The variations are classified as follows:
- Administrative changes
- Changes to the quality part of the dossier
- Changes to the safety, efficacy and pharmacovigilance part of the dossier
- Changes to the vaccine antigen master file (VAMF) part of the dossier
VNRAs will be processed as follows:
- The MAH will:
– Record the change in the Product Union Database (UPD) within 30 days of implementation including required documents (no application form is necessary).
– Documents submitted directly to UPD. No CESP submission. Documents include those listed in the Implementing Act as well as SPC, package leaflet, labels.
- The relevant CA/RMS will
– Approve/reject the variation
– Inform MAH & CMS by recording decision in database and by e-mail
CMDv has written a Best Practice Guide for variations not requiring assessment in order to provide detailed guidance on the new process.
The EMA website includes a video tutorial showing how to submit a VNRA via the Union Product Database here.
Variations Requiring Assessment (VRA)
Every change not listed in the Implementing act mentioned above (2021/17)* will require a variation that needs to be assessed.
CMDv and EMA have written a new classification guideline for the VRAs:
Guidance on the details of the classification of variations requiring assessment according to Article 62 of Regulation (EMA/CMDv/7381/2021).
The format and categorisation is similar to the previous regulation which applied (Commission Regulation 1234/2008 ), however there are many differences.
The variations are divided into chapters as follows:
E. Administrative changes
F. Quality changes
G. Safety, Efficacy and Pharmacovigilance changes
H. VAMF or, PTMF changes
I. Changes of active substance(s), strength, pharmaceutical form, route of administration or food producing target species
Z-categories have also been included to address unlisted variations and VNRA, if requirements laid down in the Implementing Regulation are not met.
The timetable for VRAs is also outlined in the new guidance as follows:
- a standard timetable, denoted by ‘S’ which will be 60 days
- a reduced timetable, denoted by ‘R’ which will be 30 days
- an extended timetable, denoted by ‘E’ which will be 90 days
For details on how to submit a VRA, CMDv have written a Best Practice Guide for Variations Requiring Assessment (EMA/CMDv/144277/2021). It has been prepared in order to facilitate the processing of VRAs for MRP/DCP products. The same general principles will apply to purely nationally authorised products.
Recommendation for the classification of variations not already listed
A procedure for requesting a recommendation for the classification of variations not already listed in either the above-mentioned Implementing Regulation or the EMA/CMDv Guidance on variations requiring assessment has also been established. This is similar to the previous CMDv recommendations for classification of unforeseen variations, according to Article 5 of Regulation 1234/2008.
Refer to CMDv new guideline for detailed advice this new process entitled: Procedural advice for requests for the classification of variations not already listed in Commission Implementing Regulation (EU) 2021/17 or EMA/CMDv Guidance on the details of the classification of variations requiring assessment according to Article 62 of Regulation 2019/6 (EMA/CMDv/144284/2021).
Worksharing and Grouping
Grouping and worksharing procedures do not apply to VNRA, they only apply to VRA.
As a consequence, no VNRAs can be included in a grouping or worksharing even if they are consequential or related to the VRAs included in the grouping or worksharing procedure.
However, the introduction section of CMDv Best Practice Guide for Variations Requiring Assessment (which also covers grouping), outlines the different approaches to follow when there is a need to co-ordinate changes that are related or consequential but are classified as VNRA and VRA.
The worksharing procedure is outlined in Article 65 of the NVR and it will be compulsory to follow this procedure, when the same change is being applied in different member states. Information related to worksharing is also mentioned in the CMDv BPG for Variations Requiring Assessment. However a specific guide on worksharing has also been written by CMDv: Best Practice Guide for Worksharing (EMA/CMDv/204024/2021).
Union Product Database
Due to the new regulation, the EMA has introduced new IT systems. The main one will be the Union Product Database (UPD).
It will contain information for all authorised veterinary medicines in the EU (including all nationally authorised products). For MAHs, it will provide self-service access for specific regulatory activities, including the management of variations that do not require assessment.
For more information on implementation, training, registration and access of the UPD, refer to the following link here on the EMA website.
The UPD will be linked to the other 3 other databases in the future. These databases are at different stages of development and introduction:
- Union Pharmacovigilance Database
On 28 January 2022, the Union Pharmacovigilance Database (EVV) was successfully released. User guidance and the release notes are available here.
- Manufacturing and Wholesale Distribution Database
The Manufacturers and Wholesale Distributors database (MWD) was released on 28 January 2022. The system is an enhanced and upgraded version of EudraGMDP, the EU database of manufacturing authorisations and certificates of good manufacturing practice, with changes affecting both the veterinary and the human domains. The MWD Project Group has also adopted requirements for aligning the GDP module with the change made to the system so far. Changes to the module will be delivered in a subsequent release scheduled for Q1 2022. In addition, enhanced search facilities on the GMP module will be delivered in the same release.
- Database for the Collection of Data on Sales and Use of Antimicrobials in Animals.
IT development on the Collection of Antimicrobials Sales and Use Data (ASU) project started in January 2022. Information on the progress of this project will be published on the EMA website as this project develops.
Q&A on transitional arrangements
CMDv has prepared a Q&A document in order to assist both MAHs and NCAs in the management of the transition between the requirements of Directive 2001/82/EC and Regulation (EU) 2019/6. This Q&A document will be regularly updated and can be found under the following link.
This document includes an Annex which outlines how individual Member States will handle renewals of marketing authorisations after 28 January 2022.
Should you need any support with Veterinary Procedures feel free to contact us & the Ivowen team will be here to help.
Written by Claire Brown

